実行手順
'cg_exe' - 共役勾配法による対角化を用いたバンド計算
 ここでは例題を上げてFPSEID21コードの実行手順を説明します。ここでの手順書はすでに実行モジュールを手に入れた人が対象です。まだ 'cg_exe', 'sd_exe, 'tddft_exe'を入手していない利用者はコンパイル手順へ戻ってください。
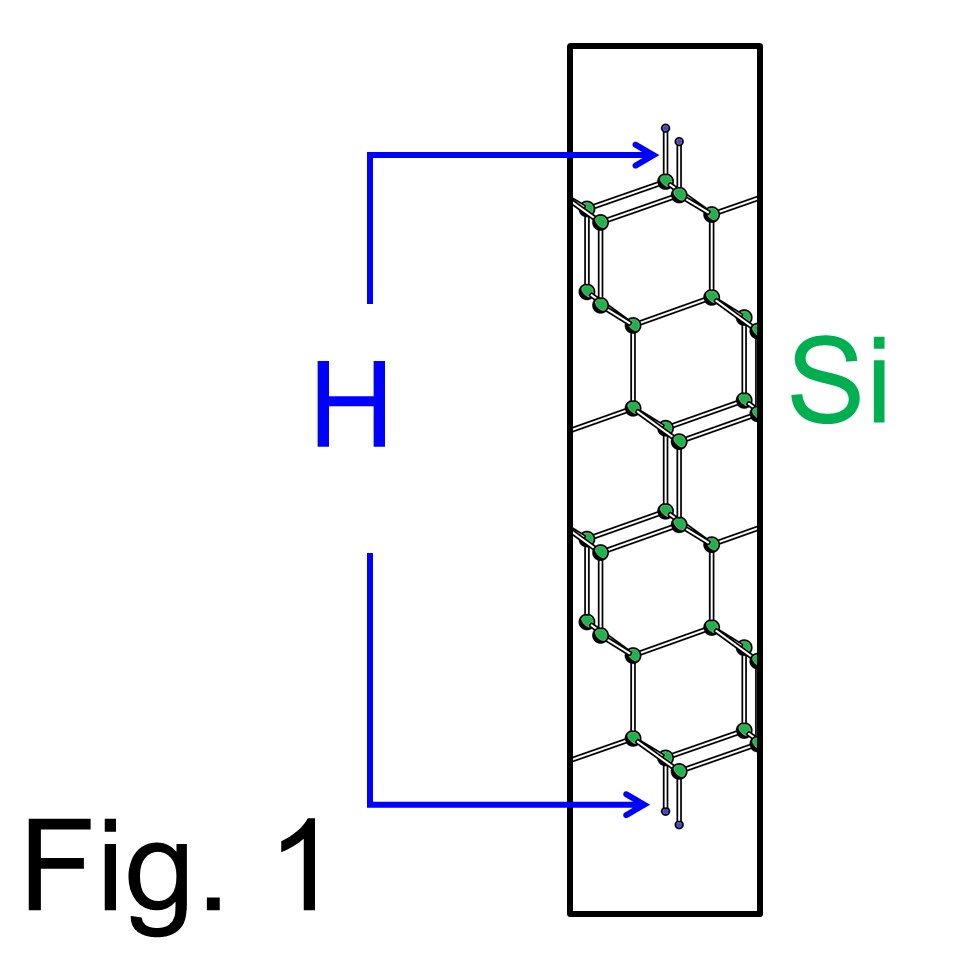
ここでは例題を上げてFPSEID21コードの実行手順を説明します。ここでの手順書はすでに実行モジュールを手に入れた人が対象です。まだ 'cg_exe', 'sd_exe, 'tddft_exe'を入手していない利用者はコンパイル手順へ戻ってください。例題の原子構造は Fig. 1で示されています。これはH終端されたSi(111)スラブモデルでスラブの上下がH原子で終端されています。ここでは通常のバンド計算によりTDDFT計算のための第一ステップを行います。TDDFT計算ではH原子のレーザー脱離をシミュレーションします。
ではいよいよTDDFT計算の'第一段階'である'cg_exe'コードの実行手順です。 Fig. 1の構造は表面平衡方向に1x1の周期をもったSi原子12層ぶんのスラブモデルです。ユニットセルの形状はhexagonalになります。では、入力データを 'Si111-H.in'と命名します。その内容は以下の boxに記載してあります。 ここでユーザーが注意する(実際に指定する)パラメーターを太字で掲載しました。 それらの詳細な意味はboxの下の本文で説明します。太字でないパラメーターは、そのままにしていただくことをお勧めします。(この入力テンプレートは 'Si111-H.in'を入手をクリックしてください。)
%%% Si(111)1x1 H-term cell %%%%
STND KPOINT=MESH ALAT=4.4427103214141699 ZVAL=50 RCUT=845 GCUT=12.0
PP=(KB,SOFT) CD=ATOM WF=INIT CG=(5,8,3) SCF=60 EXT=PANDEY
OUTWF=23 METAL=(25,0) KCONT=0 FFTWF
RMIX=0.03 CONV=1.D-7 MAXFN=0 OKSTEP=0.1D0/
1.4142135623730951 -0.8164965809277260 0.0000000000000000 A1 vector
1.4142135623730951 0.8164965809277260 0.0000000000000000 A2 vector
0.0000000000000000 0.0000000000000000 13.1042541360369356 A3 vector
2 Si12H2 # of Atomic types, chemical composition
12 2 28.0855 # of type 1 atoms, # of pseudo orbitals, atomic mass
0.0000000000000000 0.0000000000000000 0.0380725996019826 TAU( 1)
0.3333333333333333 0.3333333333333333 0.0628830683197726 TAU( 2)
0.3333333333333333 0.3333333333333333 0.1388370510257218 TAU( 3)
-0.3333333333333333 -0.3333333333333333 0.1638618412213989 TAU( 4)
-0.3333333333333333 -0.3333333333333333 0.2398223878715089 TAU( 5)
-0.0000000000000000 -0.0000000000000000 0.2642651909875856 TAU( 6)
-0.0000000000000000 -0.0000000000000000 -0.0380725996019826 TAU( 7)
-0.3333333333333333 -0.3333333333333333 -0.0628830683197726 TAU( 8)
-0.3333333333333333 -0.3333333333333333 -0.1388370510257218 TAU( 9)
0.3333333333333333 0.3333333333333333 -0.1638618412213989 TAU( 10)
0.3333333333333333 0.3333333333333333 -0.2398223878715089 TAU( 11)
0.0000000000000000 0.0000000000000000 -0.2642651909875856 TAU( 12)
-2 0 1.0 # of type 2 atoms (negative!), # of pseudo orbitals, atomic mass
-0.0000000000000000 -0.0000000000000000 0.3131888120278811 TAU( 13)
0.0000000000000000 0.0000000000000000 -0.3131888120278811 TAU( 14)
30.0 Rcut for LVGENX
4 total # of k-point mesh in one direction
1 1 0 starting points of k-point mesh for B1 B2 B3 vectors
2 2 4 skipping # of k-point mesh for B1 B2 B3 vectors
1 0 0 just these three lines
0 1 0
0 0 1
0 0 0
10 10 10 NDX NDY NDZ for DOS
2.0 2.0 for type 1 -- #s of electrons on s- and p-orbitals
1.0 0.0 for type 2 -- #s of electrons on s- and p-orbitals
最初の行の Si(111)1x1 H-term cell は計算対象のタイトルです。これは任意の形式でよいです。
2行目からヘッダーの最後までは
同じく2行目の ZVAL=50 はユニットセル当たりの全価電子数です。 Fig. 1の系はSi原子12個、H原子2個を含みますのでユニットセル当たりの全価電子数は= 4x12 + 2 =50です。 ( FPSEID21 コードはスピン分極を入れない近似のもと、価電子のみを直接扱います。) RCUT=845 はEwald和に必要な値で、ユニットセルの格子ベクトルの中で最も長いものの2乗(単位はBohr半径の2乗になります)より大きめに指定してください。GCUT=12.0は電荷密度とポテンシャルを生成する際の平面波基底のカットオフエネルギーで単位はRydbergになります。
3行目の CD=ATOM とは計算が電荷密度の初期予想としてユニットセル中の各原子の擬ポテンシャル計算で用いるpseudo-orbitalによる電荷密度の重ね合わせを与えることを意味します。(ノルム保存擬ポテンシャルの情報は実行スクリプトの中で入力ファイルより読み取るように指定されます。擬ポテンシャルの利用はのちに詳細を説明します。.) WF=INIT とは計算は価電子の波動関数の初期予想から始めることを意味し最初は各準位の波動関数当たり一種類のGベクトルによる平面波を当てはいます。 (もし、この計算が依然行われたものの繰り返しですでに電子密度と波動関数ができている安倍には CD=FILE WF=FILEと記載します。)この行のCGと書かれたラベルは大変重要で、のちに説明する 'sd_exe' 実行時には変更します。.
4行目の METAL=(25,0) は占有されたバンド数が25で、金属的バンド数が0ということです。 (金属的バンドとはFermi準位をよぎるバンドのことです) FPSEID21パッケージでは価電子のみが直接取り扱われスピン分極を無視する近似を行います。Fig. 1の例では12 Si原子と2 H原子がいますので、ユニットセル当たりの全価電子数 = 4x12 + 2 =50となります。ですので、スピン当たりの全占有バンド数は25になります。(ユーザーの方の中に金属的な系の計算にご興味の方がおられると思います。金属的な系の計算にはいくつかのトリックが必要なので、どうしても金属系の計算をご希望の方は お問い合わせまで。) 同じ行の FFTWF は電子の波動関数を計算終了時にG-空間からr実空間へフーリエ変換することを指定します。実空間の波動関数は 'sd_exe'の入力となり、波動関数をG-空間のフルグリッドへ拡張するのに用いられます。
5行目のCONV=1.D-7はセルフコンシステントポテンシャルの収束判定値です。(この値は 'tddft_exe'を実行する際には時間発展計算の高効率化のために1.D-5に変更されます。)この行は'/'文字で終了していますが、これは入力のヘッダー部分の終了を意味します。
ヘッドラインが終わりその直下の3行は格子ベクトル(A1-A3 vectors)を意味します。これらはCartesian座標で入力されます。全格子ベクトルの(x,y,z)要素は計算の中でALATの値が掛けられます。 注意! A1-A3ベクトルは左手系で与えてください。A1ベクトルは左手親指、A2ベクトルは左手中指、そしてA3ベクトルは左手人差し指に対応します。もし、A1-A3ベクトルを右手系で与えた場合には、計算の正しい実行は保証されません。
格子ベクトルの直下に'2 Si12H2'と記載があります。コードが実際に読み込むのは '2'だけで、これはユニットセル中に何種類の原子がいるかを指定します。今の例では2種類の原子 (SiとH)がいますので'2'です。
次の行はSi原子の説明です。12 2 28.0855はそれぞれユニットセル内のSi原子の数、ノルム保存擬ポテンシャルの参照するpseudo-orbitalの数、それと重さを指定します。原子の重さはプロトンの重さを単位に指定します。 今回の例のSiの場合擬ポテンシャルはSi 3s および 3p 成分を含みますので、対応するpseudo-orbitalの数は2を指定します。 (ちなみにCu,の場合ではpseudo-orbitalsの数は3で、 Cu 4s, 4p, そして 3d 成分がポテンシャルに考慮されます。その一方で,C原子の場合には、擬ポテンシャルの参照するpseudo-orbitalsの数は1のみで C 2s軌道が参照されます。C 2p成分は電荷の初期予想にのみ使用されます。) それに続く 12行は原子座標でA1-A3 ベクトルを単位とします。このような原子座標の指定は他のDFTコードでもよく見られると思います。
次の行は H 原子の記述です。 -2 0 1.0 はそれぞれユニットセル当たりのH原子の数、 pseudo-orbitalの数、そして電子質量です。(同位体分布を無視して1.0にしています)水素原子の数に限りマイナスの値を指定するのは奇妙ですが、これはHやHeのようにpseudo-orbitalを擬ポテンシャルの参照軌道にしない特殊なケースに限られています。歴史的に擬ポテンシャルの数 '0'とリンクしてその元素の数をあえて負の値で与える仕様になっていることご理解ください。正の値とpseudo-orbitalの数'0'の組み合わせがうまく働くかはチェックされていません。続いての2行は H 原子の座標でこれも A1-A3 ベクトルを単位としています.
原子座標の後に続く行は、計算で用いられるk-点メッシュを指定しています。
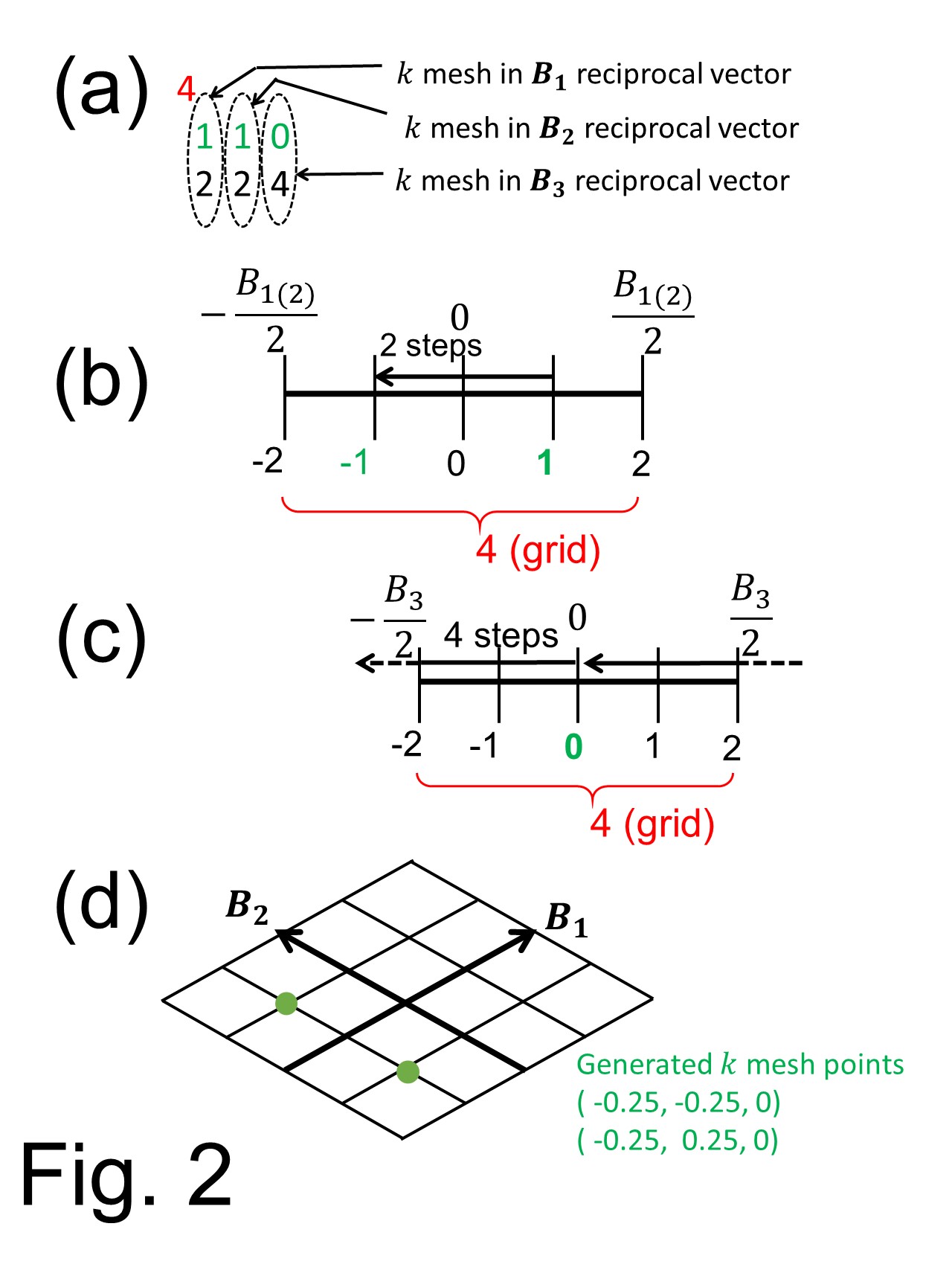 上のBox内の入力データ'Si(111)-H.in'では次のような数列があります。
上のBox内の入力データ'Si(111)-H.in'では次のような数列があります。 4
1 1 0
2 2 4
一番上の数 4はk-ベクトルのグリッド数ですべての逆格子ベクトル B1 B2 B3 で共通です。この例では B1 B2 B3 ベクトルは)グリッド点を-2から2まで与えられます。(Fig.2 (b)2番目の行 1 1 0はB1-B3各ベクトル上で k-点メッシュを発生させる始点をあたえ、それぞれの番号はその直下の番号と関係があります。( Fig. 2 (a)参照 ). 3番目の行 2 2 4 はあるgrid点から隣のgrid点へ移動する際のstep数を表します。( 今回の例では B1 B2 ベクトルの場合はFig. 2 (b)を参照、そして B3 vectorの場合はFig. 2 (c)を参照してください。)このようは手順の場合B1-B3各ベクトルの方向に作成されるk点は緑の番号で示されます。 最終的に各逆格子ベクトル上のk点のgrid点を用いて第一ブリルアンゾーン中のk点が作られます。 Fig. 2 (d)はそうして得られた最終のk点でB3ベクトル方向にはk=0だけがとられ2次元グリッドとなります。ここでスピン軌道相互作用を無視した近似を行っていますのでk 点と-k 点のBloch状態は複素共役縮退をしています。なので縮約された k点の数は2となります。
上のBox内の最後の2行はSiとHそれぞれの原子の価電子軌道への電子占有を示します。 Si 2sと2p軌道はそれぞれ 2電子に占有され、 H 1s 軌道と 2p 軌道はそれぞれ 1 、 0 電子に占有されています。 (またまた H 原子の場合は奇妙かもしれませんが、今採用している擬ポテンシャルは同じ角運動量のpseudo-orbitalを参照しませんので1sと2p軌道の占有がここで指定されます。実際に2p軌道の占有は0なので電荷密度の初期予想に影響はありません) 今回は電子軌道の占有が軌道準位の低い順に指定されますが、Cu原子の場合には軌道準位の順番とはなりません。Cu 原子は価電子の軌道として3d. 4s (そして4p)軌道を考えますが、電子占有を指定する際には 1.0 0.0 10.0と書くのが、FPSEID21の擬ポテンシャルの様式とマッチします。つまり 4s, 4p 3d の順番であり、軌道準位の順番ではなく軌道角運動量の順番になる規則であることをご了承ください。
上記の入力ファイル以外の入力ファイルに 'size.dat'と 'sym.C1' および擬ポテンシャルのファイルがあります。これらはみなアスキー形式ですがFPSEID21 パッケージに特化した内容となります。.
まず 'size.dat'はフリーフォーマットのアスキー形式で、 FPSEID21パッケージコードの計算実行の際に必要な変数配列の長さの情報を与えます。その数値は以下に説明する手順でえることができ、今回の例題に沿うとその内容は以下になります。
15 15 129 for FFT mesh grids 実空間FFTメッシュ数
18628 2328 for NG and NG2 : G for potential and wf
2 # of irreducible k-points (縮約されたk点の数)
25 7 # of occpied and empty bands (占有、空の軌道数)
14 2 # of atoms and atomic types per cell (原子種の数と総原子数)
rGcut=sqrt(Gcut)
Gnum=(2*rGcut)**3*Vol/(6*pi**2) * 1.25d0
Gnum2=Gnum/8.d0
NG=Int(Gnum)
NG2=Int(Gnum2)
この式の 'rGcut'は入力データGUCT=で示されたカットオフエネルギーです。(単位はRydberg) 'Vol' はユニットセルの体積で Bohr3の単位です。
平面波基底に用いるFFT 計算のグリッド数 (NRX, NRY, NRZ) 以下の式で決定します。
A1leng=ALAT*sqrt{ A1(1)**2 + A1(2)**2 + A1(3)**2 )
NRX=int(2*A1leng*Gcut/pi+1.d0)
A2leng=ALAT*sqrt{ A2(1)**2 + A2(2)**2 + A2(3)**2 )
NRY=int(2*A2leng*Gcut/pi+1.d0)
A3leng=ALAT*sqrt{ A3(1)**2 + A3(2)**2 + A3(3)**2 )
NRZ=int(2*A1leng*Gcut/pi+1.d0)
そのあとで NRX NRY NRZ 得られた値に近い奇数の整数に選択しなおします。この際にFFTの性能が良くなるように素数は選択しないことが大事です。
一方ファイル 'sym.C1'は計算対象の対称性がないという指定をします。これは単位行列の情報が入っていますが書式が FPSEID21向けに厳格に決まってしまっていますので、このファイルをsym.C1からダウンロードしていただくことを推奨します。
最後に必要な入力ファイルは擬ポテンシャルですが、それは実行スクリプトの説明と共に行います。
以下に示しますのは、 'cg_exe'の実行スクリプトの例です。これは、利用者の方のLinux環境に依存します。 まずファイル指定を示すFORT41, FORT46, FORT42の行を見てください。 (NECのAuroraのユーザーはVE_FORT41, VE_FORT46, VE_FORT42のようにファイル指定を書き換えてください) これらはノルム保存型擬ポテンシャルで次の文献の方法で計算されています。 [N. Troullier and J. L. Martins, 'Efficient pseudopotentials for plane-wave calculation', Phys. Rev. B43, 1993 (1991)]. これらの擬ポテンシャルはアスキー形式でbox中に示したスクリプトからダウンロード可能です。 ここで示したスクリプトと整合性をとるために、ユーザーの方には自分のホームディレクトリーに'TR'というディレクトリーを作成いただき、ダウンロードした擬ポテンシャルを'TR'へ格納していただくことを推奨します。i番目の原子種(注:i番目の原子座標ではありませn)に対応する擬ポテンシャルファイルI/O番号は40+iになります。これは軽元素(H - Ne)に対応します。一方で、より重い種類の元素の擬ポテンシャルは2つのファイルに分かれていてファイル番号は40+iと40+i+5になります。 これはFPSEID21特有の形式であることにご留意ください。この場合に40+i番に指定する擬ポテンシャルのフェイル名は名前の最後に'g.asci' とか 'g.asc'という文字が付きます。一方で、40+i+5のファイル番号に対応する擬ポテンシャルファイルは名前の終わりに 'e.asci' とか 'e.asc'という文字が来ます。以下のboxに書いてあるスクリプトでこのルールを確認してください。以下のスクリプトより、今回の例題に用いるSiとHの擬ポテンシャルをスクリプト中に示されたファイル名をクリックするとダウンロードすることができます。
#$ -N cg_exe_Si111-H-1x1H
#$ -S /bin/bash
#$ -j y
#$ -e /home/youraccount/Si111-H/std_omp.err
#$ -o /home/youraccount/Si111-H/std_omp.out
#$ -pe mpi 16
export DIR=/home/youraccount
export DIR2=/home/youraccount/Si111-H
### --- input files
### pseudopotentials
export FORT41=$DIR/TR/TR.Si93g_asci ! Si Pseudopotentials (g)
export FORT46=$DIR/TR/TR.Si93e_asci ! Si Pseudopotentials (e)
export FORT42=$DIR/TR/TR.H99g_asc ! H Psedutopotentials
###
export FORT54=$DIR2/size.dat
export FORT55=$DIR2/sym.C1
###
export FORT20=$DIR2/rh.Si111-H ! input charge when 'CD=FILE' in 'Si111-H.in'
export FORT22=$DIR2/wf.Si111-H ! input wavefunction when 'WF=FILE' in 'Si111-H.in
### --- output files
export FORT23=$DIR2/wf.Si111-H_new ! output wavefunction (G-spave compact sphere)
export FORT88=$DIR2/wf_real.Si111-H ! output wavefunction (real space) used by 'sd_exe'
export FORT90=$DIR2/Vall.Si111-H
export FORT24=$DIR2/rh.Si111-H_new ! output charge density
export FORT25=$DIR2/Vint.temp
export FORT77=$DIR2/tau.Si111-H_new ! atomic coordinate stored at the end of 'cg_exe'.
export FORT78=$DIR2/need
export OMP_NUM_THREADS=4
cd $DIR2
$DIR/lm/cg_exe < Si111-H.in > Si111-H.out
'cg_exe' が実行されますとバイナリー形式の出力ファイル 'rh.Si111-H_new'、'wf.Si111-H_new'、'wf_real.Si111-H_new' が生成されるのを確認してください。続いて 'rh.Si111-H_new'を 'rh.Si111-H'にファイル名を変更し、'wf.Si111-H_new'、'wf_real.Si111-H_new'をそれぞれ'wf.Si111-H'、'wf_real.Si111-H'に変更してください。続いて実行出力ファイル'Si111-H.out'内でSCF収束していることを確認してください。そのためには
$ grep ITR Si111-H.out
コマンドによる出力を見て、収束判定条件を満たしていることを確認してください。収束が不十分であれば、'cg_exe'をもう一回実行しますが、その際には入力ファイル'Si111-H.in'のヘッダー部のCD=ATOMをCD=FILEに、WF=INITをWF=FILEに変更してから実行してください。収束判定条件を確認したら、再び'*_new'という名のファイル全てを最後の文字列'_new'のないファイル名へと上書きしてください。
次のステップは 'sd_exe'の実行です。 'sd_exe'の実行には 'sd_exe'の実行手順へ進んでください。
また'cg_exe'計算結果の標準出力ファイル 'Si111-H.out' (アスキー形式)は上のboxの該当するか所からダウンロードできます。以下は出力ファイルの中の主要な部分の説明で赤い文字で示してあります。
-------
This is CG-code ...
-------
%%% Si(111)1x1 H-term cell %%%%
# OF COMMANDS = 20
NO. 1 COMMAND = STND OPERATION =
NO. 2 COMMAND = KPOINT OPERATION = MESH
NO. 3 COMMAND = ALAT OPERATION = 4.4427103214141699
- - - - - - - - - - -
- - - - - - - - - - -
NO. 19 COMMAND = MAXFN OPERATION = 0
NO. 20 COMMAND = OKSTEP OPERATION = 0.1D0
- - - - - - 中略 - - - - -
INITIAL POSITION OF ATOM IN
1 0.0000000 0.0000000 2.2165260
2 4.1886275 0.0000000 3.6609519
3 4.1886275 0.0000000 8.0828717
4 -4.1886275 0.0000000 9.5397751
- - - 中略 - - -
OMEGA= 2653.7234450
Read S-matrices fron file !!
# OF SYMMETRY OPERATIONS: NTOT = 1
S MATRICES: OP 1ST RAW 2ND RAW 3RD RAW
1 1 0 0 0 1 0 0 0 1
- - - - - 中略 - - - -
A-VECTORS
6.2829412 6.2829412 0.0000000
-3.6274578 3.6274578 0.0000000
0.0000000 0.0000000 58.2184051
B-VECTORS
0.3535534 0.3535534 0.0000000
-0.6123724 0.6123724 0.0000000
0.0000000 0.0000000 0.0763111
- - - - 中略 - - - -
**** MESHK: N = 4
TOTAL NO. OF K IN WHOLE BZ = 4
TOTAL NO. OF K IN WEDGE = 2
NO. COORDINATES WK
1 -0.2500 -0.2500 0.0000 1.0000
2 -0.2500 0.2500 0.0000 1.0000
- - - - 中略 ----
*** ITR # 1 CONVERGENCE OF THE POTENTIAL IS ** 0.1365134D+00
*** ITR # 2 CONVERGENCE OF THE POTENTIAL IS ** 0.4149469D-01
- - - -
*** ITR # 23 CONVERGENCE OF THE POTENTIAL IS ** 0.2281881D-06
*** ITR # 24 CONVERGENCE OF THE POTENTIAL IS ** 0.9713493D-07
Store SCF local potential
Fourier components of eigen values
Fourier components of occupation #s
( 'EIG'の真下のカラムが固有値).
IB
EIG
CONV'
SIN(ROT)
OCC
K VECTOR 1 -0.1767767 0.0000000 0.0000000 CARTESIAN
BAND (EV) 1 -0.1197297E+02 0.1811632E-07 0.1501256E-03 1.000
BAND (EV) 2 -0.1175938E+02 0.1691344E-07 0.1520060E-03 1.000
BAND (EV) 3 -0.1141360E+02 0.2046485E-07 0.1657217E-03 1.000
- - - - 中略 - - -
K VECTOR 2 0.0000000 0.3061862 0.0000000 CARTESIAN
BAND (EV) 1 -0.1063678E+02 0.1632811E-08 0.1054169E-03 1.000
BAND (EV) 2 -0.1044811E+02 0.1177430E-08 0.1039971E-03 1.000
BAND (EV) 3 -0.1014525E+02 0.1802020E-08 0.1063807E-03 1.000
- - - - 中略 - - -
****** TOTAL ENERGY: ETOT = -0.4867975154D+02 HR
-0.9735950309D+02 RYD
******
ESELF = 0.5052927104D+01 HR
******
EWA = 0.1842936419D+03 HR
******
ELOCAL = -0.4850683943D+03 HR
******
EXC = -0.1499663146D+02 HR
******
EH = 0.2317450508D+03 HR
******
EKINE = 0.1899942511D+02 HR
******
ENL = 0.1129422925D+02 HR
****** TOTAL FORCE: NEGATIVE (HARTREE/AU):
1 -0.0000203 0.0000032 0.0014520
2 0.0001712 0.0000095 -0.0002048
3 -0.0000624 -0.0000051 -0.0000924
- - - skipping - - -
13 -0.0005073 0.0000030 -0.0005740
14 0.0005073 -0.0000030 0.0005740