実行手順
'sd_exe' - 再急降下法による対角化を利用したバンド計算
 これは 'sd_exe' の実行手順を示すマニュアルで、例題は同じくFig 1の系です。 この案内はすでに 'cg_exeを実行済みの利用者向けですので、、まだ 'cg_exe'を実行済みでない方は'cg_exe'の実行手順へお戻りください。 (実行モジュール 'cg_exe'、 'sd_exe、 and 'tddft_exe'のどれか一つでも作成しておられない利用者の方はコンパイル手順へ戻ってください。)
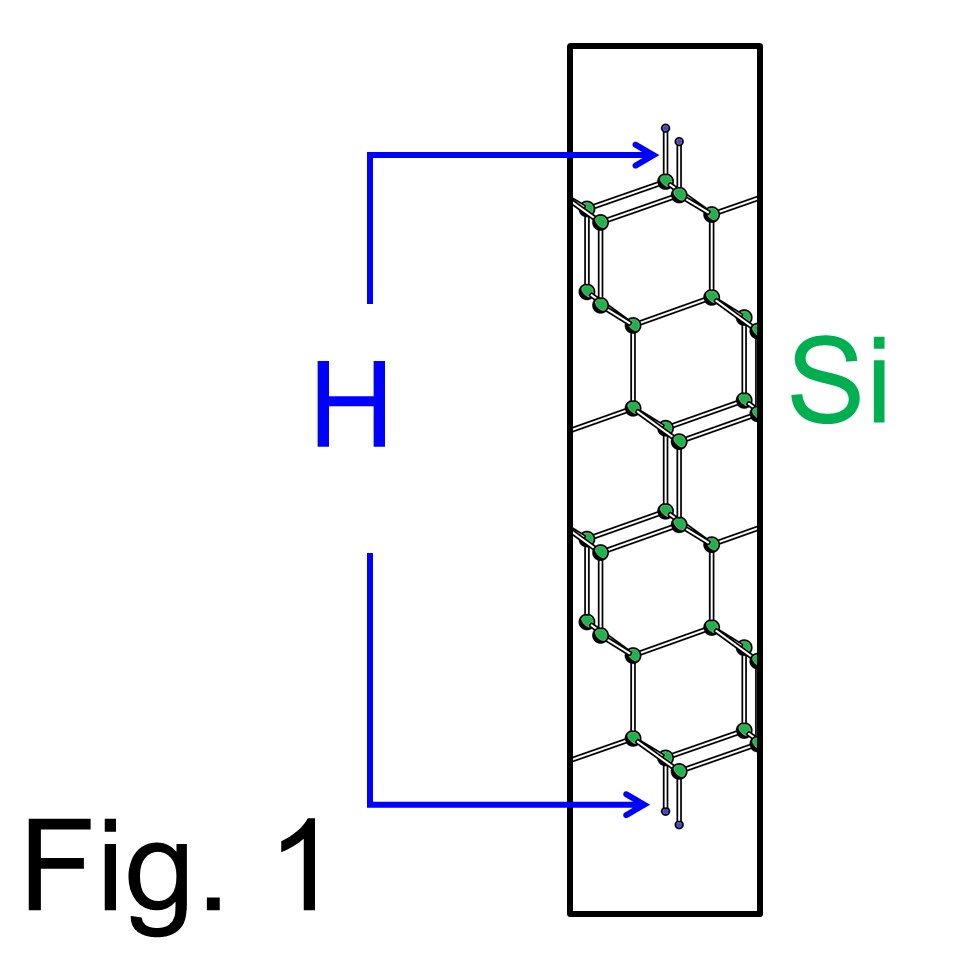
これは 'sd_exe' の実行手順を示すマニュアルで、例題は同じくFig 1の系です。 この案内はすでに 'cg_exeを実行済みの利用者向けですので、、まだ 'cg_exe'を実行済みでない方は'cg_exe'の実行手順へお戻りください。 (実行モジュール 'cg_exe'、 'sd_exe、 and 'tddft_exe'のどれか一つでも作成しておられない利用者の方はコンパイル手順へ戻ってください。)例題は 'cg_exe'実行時と同じで Fig. 1のH原子終端されたSi(111)スラブモデルで、上端と下端に水素原子が吸着しています。 ここでは、バンド計算を実行することにより、通常のバンド計算結果からG-spaceのフルグリッドで価電子の波動関数を得ることを行います。これはTDDFT計算のための2ステップ目の準備計算で、TDDFT計算ではFig.1の構造においてレーザー照射によるH原子脱離のシミュレーションを行います。
Fig. 1の構造は'cg_exe'実行時と同じですので、入力データは少し変更するだけで十分です。変更部分を以下のBox内で示します。変更は少しですが 'sd_exe'実行には非常に重要です。まず CD=ATOM は CD=FILEに変更、 WF=INIT は WF=FFTに変更してください。さらに FFTWFの文字は削除です。そして次の文字は CG から SDに変更してください。変更部分は色付きで強調し、そのほかの部分は変更してはいけません。(以下のboxに示した内容は download 'Si111-H_sd.in'をダウンロードからダウンロードできます。)
%%% Si(111)1x1 H-term cell %%%%
STND KPOINT=MESH ALAT=4.4427103214141699 ZVAL=50 RCUT=845 GCUT=12.0
PP=(KB,SOFT) CD=
OUTWF=23 METAL=(25,0) KCONT=0
RMIX=0.03 CONV=1.D-7 MAXFN=0 OKSTEP=0.1D0/
1.4142135623730951 -0.8164965809277260 0.0000000000000000 A1 vector
1.4142135623730951 0.8164965809277260 0.0000000000000000 A2 vector
0.0000000000000000 0.0000000000000000 13.1042541360369356 A3 vector
2 Si12H2 # of Atomic types, chemical composition
12 2 28.0855 # of type 1 atoms, # of pseudo orbitals, atomic mass
0.0000000000000000 0.0000000000000000 0.0380725996019826 TAU( 1)
0.3333333333333333 0.3333333333333333 0.0628830683197726 TAU( 2)
0.3333333333333333 0.3333333333333333 0.1388370510257218 TAU( 3)
-0.3333333333333333 -0.3333333333333333 0.1638618412213989 TAU( 4)
-0.3333333333333333 -0.3333333333333333 0.2398223878715089 TAU( 5)
-0.0000000000000000 -0.0000000000000000 0.2642651909875856 TAU( 6)
-0.0000000000000000 -0.0000000000000000 -0.0380725996019826 TAU( 7)
-0.3333333333333333 -0.3333333333333333 -0.0628830683197726 TAU( 8)
-0.3333333333333333 -0.3333333333333333 -0.1388370510257218 TAU( 9)
0.3333333333333333 0.3333333333333333 -0.1638618412213989 TAU( 10)
0.3333333333333333 0.3333333333333333 -0.2398223878715089 TAU( 11)
0.0000000000000000 0.0000000000000000 -0.2642651909875856 TAU( 12)
-2 0 1.0 # of type 2 atoms (negative!), # of pseudo orbitals, atomic mass
-0.0000000000000000 -0.0000000000000000 0.3131888120278811 TAU( 13)
0.0000000000000000 0.0000000000000000 -0.3131888120278811 TAU( 14)
30.0 Rcut for LVGENX
4 total # of k-point mesh in one direction
1 1 0 starting points of k-point mesh for B1 B2 B3 vectors
2 2 4 skipping # of k-point mesh for B1 B2 B3 vectors
1 0 0 just these three lines
0 1 0
0 0 1
0 0 0
10 10 10 NDX NDY NDZ for DOS
2.0 2.0 for type 1 -- #s of electrons on s- and p-orbitals
1.0 0.0 for type 2 -- #s of electrons on s- and p-orbitals
'cg_exe'の入力データと同様
ほかの入力ファイル 'size.dat'、 'sym.C1' それと擬ポテンシャルのファイルは 'cg_exe'実行時と同じになります。 これらの入力ファイルの中身は FPSEID21 パッケージに特化した内容となっています。
ここに'sd_exe' 実行時のスクリプトを示します。'cg_exe'実行時のスクリプトからの変更は少しだけで、変更部分は以下のbox内で赤字で示します。 ファイルI/OのFORT22では該当するファイル名を 'wf.Si111-H' から 'wf_fft.Si111-H'に変えます。これはG-spaceフルグリッドでの波動関数ですが、この入力ファイルの使用はまれです。なぜなら、多くのケースでは 'sd_exe' を実空間上の波動関数から初めて実行するとSCF収束にすぐに至るからです。ファイルI/OのFORT23では該当するファイル名を 'wf.Si111-H_new'から 'wf_fft.Si111-H_new' .に変えます。これは出力される波動関数がG-spaceのフルグリッドで表現されるものにかえます。また、以下のBox内では入力ファイル名(上記Box内)を'Si111-H_sd.in'に、出力ファイル名'Si111-H_sd.out'に変更しています。それぞれの内容は、以下のbox内スクリプトの当該箇所をクリックするとダウンロードできるようになっています。
#$ -N sd_exe_Si111-H-1x1H
#$ -S /bin/bash
#$ -j y
#$ -e /home/youraccount/Si111-H/std_omp.err
#$ -o /home/youraccount/Si111-H/std_omp.out
#$ -pe mpi 16
export DIR=/home/youraccount
export DIR2=/home/youraccount/Si111-H
### --- input files
### pseudopotentials
export FORT41=$DIR/TR/TR.Si93g_asci ! Si Pseudopotentials (g)
export FORT46=$DIR/TR/TR.Si93e_asci ! Si Pseudopotentials (e)
export FORT42=$DIR/TR/TR.H99g_asc ! H Psedutopotentials
###
export FORT54=$DIR2/size.dat
export FORT55=$DIR2/sym.C1
###
export FORT20=$DIR2/rh.Si111-H ! input charge when 'CD=FILE' in 'Si111-H.in'
export FORT22=$DIR2/wf
### --- output files
export FORT23=$DIR2/wf
export FORT88=$DIR2/wf_real.Si111-H ! input wavefunction (real space) used by 'sd_exe'
export FORT90=$DIR2/Vall.Si111-H
export FORT24=$DIR2/rh.Si111-H_new ! output charge density
export FORT25=$DIR2/Vint.temp
export FORT77=$DIR2/tau.Si111-H_new ! atomic coordinate stored at the end of 'sd_exe'.
export FORT78=$DIR2/need
export OMP_NUM_THREADS=4
cd $DIR2
$DIR/lm/
SCF収束性を判断するには以下のコマンドを用いてください。
$ grep ITR Si111-H_sd.out
収束判定条件が 1.D-7.以下の値で止まっていれば収束完了です。
SCF収束が達成したら。出力ファイルのうち 'rh.Si111-H_new' と 'wf_fft.Si111-H_new' がそれぞれ電荷密度とG-spaceフルグリッドで表現された波動関数で、TDDFT計算の初期入力ファイルとなります。それぞれ名前を'_new'のついていないものに変更します。最後のステップの TDDFT計算へ行くには以下のアイコンをクリックしてください。