How to run?
Run 'cg_exe' - the band structure calculation by conjugate-gradient diagonalizationn
 Here the manual gives an example to explain how to run FPSEID21 code. This guidance is written for people who already made executable load modules. If you have not obtained 'cg_exe', 'sd_exe, and 'tddft_exe', go to How to compile?
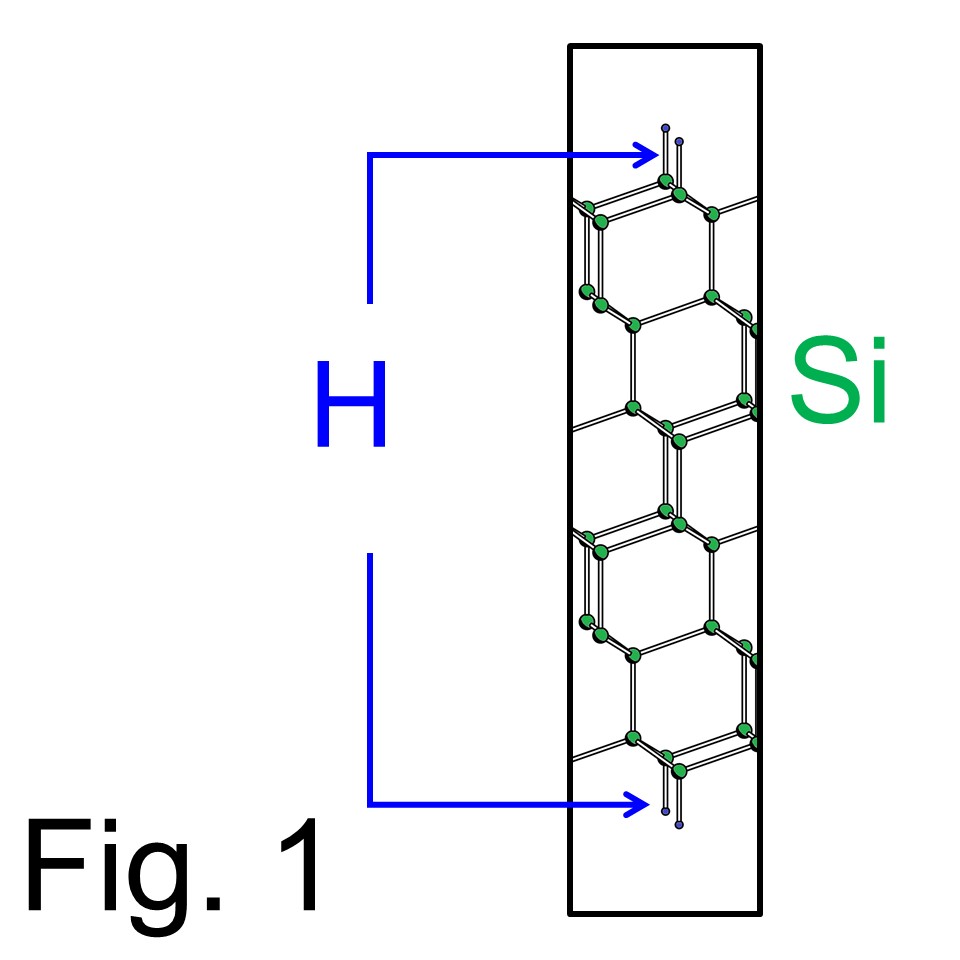
Here the manual gives an example to explain how to run FPSEID21 code. This guidance is written for people who already made executable load modules. If you have not obtained 'cg_exe', 'sd_exe, and 'tddft_exe', go to How to compile? The example has a structure as illustrated in Fig. 1, which is a Si(111) slab model terminated by H atoms on top and bottom. Here the conventional band structure calculation is performed to make first-step toward the TDDFT calculations, which simulate laser-induced H desorption.
Now this manual explains the 'first-step' running of 'cg_exe' needed for the TDDFT calculation. The structure shown in Fig. 1 has 1x1 periodicity in surface lateral directions and 12 atomic layer of Si exists in a slab. The shape of the unitcell is hexagonal. The input data named as 'Si111-H.in' is displayed inside a box below. Here, parameters requiring high attention are displayed as boldface which will be explained below the box. Other parameters should be remained as they are. (This template file is obtained from download 'Si111-H.in')
%%% Si(111)1x1 H-term cell %%%%
STND KPOINT=MESH ALAT=4.4427103214141699 ZVAL=50 RCUT=845 GCUT=12.0
PP=(KB,SOFT) CD=ATOM WF=INIT CG=(5,8,3) SCF=60 EXT=PANDEY
OUTWF=23 METAL=(25,0) KCONT=0 FFTWF
RMIX=0.03 CONV=1.D-7 MAXFN=0 OKSTEP=0.1D0/
1.4142135623730951 -0.8164965809277260 0.0000000000000000 A1 vector
1.4142135623730951 0.8164965809277260 0.0000000000000000 A2 vector
0.0000000000000000 0.0000000000000000 13.1042541360369356 A3 vector
2 Si12H2 # of Atomic types, chemical composition
12 2 28.0855 # of type 1 atoms, # of pseudo orbitals, atomic mass
0.0000000000000000 0.0000000000000000 0.0380725996019826 TAU( 1)
0.3333333333333333 0.3333333333333333 0.0628830683197726 TAU( 2)
0.3333333333333333 0.3333333333333333 0.1388370510257218 TAU( 3)
-0.3333333333333333 -0.3333333333333333 0.1638618412213989 TAU( 4)
-0.3333333333333333 -0.3333333333333333 0.2398223878715089 TAU( 5)
-0.0000000000000000 -0.0000000000000000 0.2642651909875856 TAU( 6)
-0.0000000000000000 -0.0000000000000000 -0.0380725996019826 TAU( 7)
-0.3333333333333333 -0.3333333333333333 -0.0628830683197726 TAU( 8)
-0.3333333333333333 -0.3333333333333333 -0.1388370510257218 TAU( 9)
0.3333333333333333 0.3333333333333333 -0.1638618412213989 TAU( 10)
0.3333333333333333 0.3333333333333333 -0.2398223878715089 TAU( 11)
0.0000000000000000 0.0000000000000000 -0.2642651909875856 TAU( 12)
-2 0 1.0 # of type 2 atoms (negative!), # of pseudo orbitals, atomic mass
-0.0000000000000000 -0.0000000000000000 0.3131888120278811 TAU( 13)
0.0000000000000000 0.0000000000000000 -0.3131888120278811 TAU( 14)
30.0 Rcut for LVGENX
4 total # of k-point mesh in one direction
1 1 0 starting points of k-point mesh for B1 B2 B3 vectors
2 2 4 skipping # of k-point mesh for B1 B2 B3 vectors
1 0 0 just these three lines
0 1 0
0 0 1
0 0 0
10 10 10 NDX NDY NDZ for DOS
2.0 2.0 for type 1 -- #s of electrons on s- and p-orbitals
1.0 0.0 for type 2 -- #s of electrons on s- and p-orbitals
In the first line, Si(111)1x1 H-term cell is just a title of subject. You can write anything to identify what you do.
From second line to the end of the header of the input data,
Again, in the second line, ZVAL=50 gives # of all valence electrons per unitcell. The system of Fig. 1 contains 12 Si and 2 H atoms, thus total number of valence electron per unitcell = 4x12 + 2 =50. (Note that the FPSEID21 package treats valence orbitals only within spin-unpolarized approximation.) RCUT=845 is a value needed for the Ewald summation and preferred to be longer than the longest lattice vector of the unitcell. (The unit is (Bohr radius)2) GCUT=12.0 means the cut-off energy of the plane-wave basis set to describe charge density and potential with the Rydberg unit.
In the third line, CD=ATOM means that the calculation starts with initial guess of the valence charge density which is given by atomic pseudo-orbitals. (The pseudo-orbitals is taken from a file represent the norm-conserving pseudopotentials. The usage of pseudopotentials will be explained below.) WF=INIT means that the calculation starts with initial guess of the valence wavefunctions given by plane waves with single G vector. (Please note if the calculation is done by using previously obtained charge density and wavefunctions, you should set CD=FILE WF=FILE.) The label CG in this line is necessary and should be changed when we run 'sd_exe' which will be explained later.
In the fourth line, METAL=(25,0) means, number of occupied bands is 25, and number of metallic bands is 0. (The metallic band means the bands crossing the Fermi level.) In the FPSEID21 package, only valence orbitals are treated within spin-unpolarized approximation. The system of Fig. 1 contains 12 Si and 2 H atoms, thus total number of valence electron per unitcell = 4x12 + 2 =50. Thus, the number of all occupied band per spin is 25. One can be interested in cases of metallic systems, which needs some trick and will be explained when contacted through Inquiry. In the same line, FFTWF request the Fourier transformation of wavefunction from the G-space to the real-space, that will be used to run 'sd_exe' to expand wavefunctions in full-grid G-space.
The fifth line CONV=1.D-7 gives conversion criteria of the self-consistent potentials. (This value will be increased into 1.D-5, when running 'tddft_exe' for practical efficiency. Note that this line ended with '/' which indicates the end of the header.
Below the headlines, the first three lines indicate Cartesian coordinates of lattice vectors (A1-A3 vectors). All elements of A1-A3 vectors are multiplied by a value of ALAT at running the code. Caution! The lattice vectors A1-A3 should be given in the Left-handed coordinates, in which vector A1 corresponds to your left thumb, A2 corresponds to your left middle finger, and A3 corresponds to your left index finger. If you define A1-A2 vector in the Right-handed coordinates, the correctness of the simulation is not guaranteed.
Below the lattice vectors, '2 Si12H2' is denoted. The code actually read '2' only which is a number of atomic type in the unitcell. In this example, there are two kinds of atoms (Si and H).
Next line describes Si atoms and 12 2 28.0855 respectively describe # of Si atoms, # of pseudo-orbitals for the norm-conserving pseudopotentials, and atomic mass in unit of mass of proton. In Si case, the pseudopotentials have Si 3s and 3p components, thus the # of pseudo-orbital is 2. (In case of Cu, the # of pseudo-orbitals is 3, Cu 4s, 4p, and 3d components. Meanwhile, in case of C, the # of pseudo-orbitals is 1, C 2s component. Note that the C 2p component is not used.) Then following 12 lines are atomic coordinates of Si atoms in unit of A1-A3 vectors.
Next line describes H atoms and -2 0 1.0 respectively describe # of H atoms, # of pseudo-orbitals for the norm-conserving pseudopotentials, and atomic mass in unit of mass of proton. It is strange that number of atoms is given in negative one but it is only in cases of H and He in which pseudopotentials are given as local potentials (no need to use pseudo-wavefunctions for nonlocal part). This strange rule is historically given linked with '0' for # of pseudo-orbitals and validation in using positive number has not been checked. Then following 2 lines are atomic coordinates of H atoms in unit of A1-A3 vectors.
After atomic coordinates of H atoms, the next coming lines represents k-vector mesh points used in the calculation.
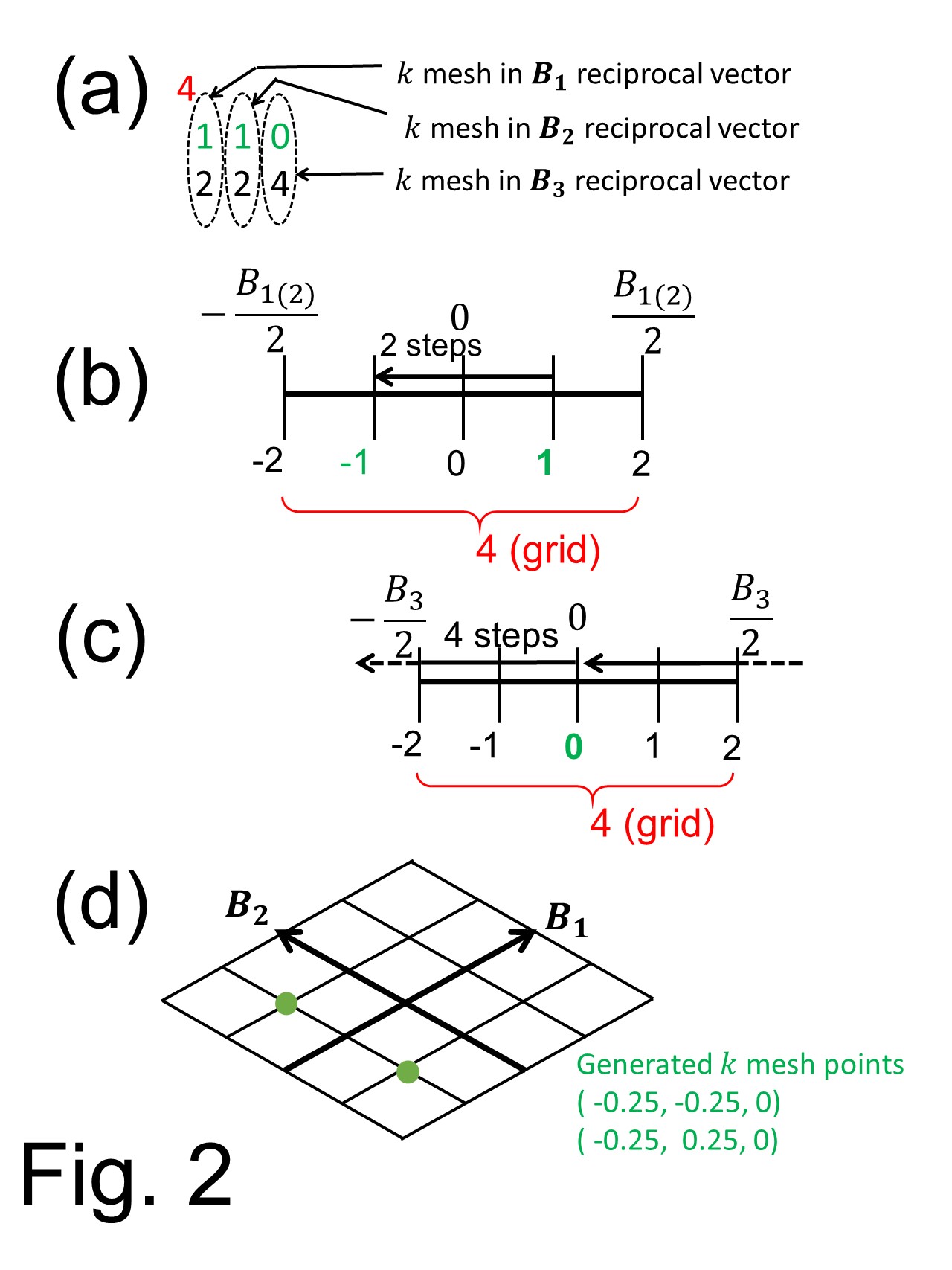 In the 'Si(111)-H.in' data shown in above box, there is
In the 'Si(111)-H.in' data shown in above box, there is 4
1 1 0
2 2 4
Here, the top number 4 defines grid # of k-vector in all reciprocal vectors. Thus, each reciprocal vector (here called as B1 B2 B3 vectors) has grid points from -2 to 2 as shown in Fig. 2 (b) to divide one of reciprocal vector into four pieces. The second line 1 1 0 denotes the starting point to generate the k-point and each number links to the number directly below, as shown in Fig. 2 (a). The third line 2 2 4 show steps to move one grid to neighbor, as shown in Fig. 2 (b) for B1 B2 vectors and in Fig 2 (c) for B3 vector. In this manner, grid point for each reciprocal vectors generated as shown in green-colored numbers in Figs. 2 (b) and (c). Finally, the generated grid points of all reciprocal vector are used to generate k-points in the first Brillouin zone. Fig. 2 (d) shows the current case in which the k point has zero component in B3 vector. Note that the current case has no spin-orbit interaction thus there is degeneracy between Block state with vector k and -k, thus the irreducible k-points is two.
The bottom two line shows electron occupation on each atomic valence orbitals for Si and H. The Si 2s and 2p orbitals respectively have 2 elecrtons, while H 1s and 2p orbitlas has 1 and 0 electrons. (The case H atom may sounds strange but currently employed pseudopotential do not referr the orbitals with the same angular momentum.) Here is not the case of Cu which has 3d. 4s (and 4p) orbitals as valence electrons. But Cu case needs to define the occupation number as 1.0 0.0 10.0 as current format orders pseudo-wavefunctions and pseudopotentials in increasing order of the angular momentum, thus 4s, 4p and 3d.
Besides the 'Si111-H.in' file, there are files for input data as 'size.dat', 'sym.C1' and pseudopotentials. All of these have specific contents suitable only to FPSEID21 package.
The 'size.dat' which is an asci file with free-format, gives necessary size of arrays used in FPSEID21 package which is genrated by following procedure.he contents of 'size.dat' for this example is as follows with explanations:
15 15 129 for FFT mesh grids
18628 2328 for NG and NG2 : G for potential and wf
2 # of irreducible k-points
25 7 # of occpied and empty bands
14 2 # of atoms and atomic types per cell
rGcut=sqrt(Gcut)
Gnum=(2*rGcut)**3*Vol/(6*pi**2) * 1.25d0
Gnum2=Gnum/8.d0
NG=Int(Gnum)
NG2=Int(Gnum2)
Here, 'rGcut' means the cutt-off energy explained above.'Vol' means the volume of the unit cell in Bohr3 unit.
The FFT grid (NRX, NRY, NRZ) are obtained as following procedure
A1leng=ALAT*sqrt{ A1(1)**2 + A1(2)**2 + A1(3)**2 )
NRX=int(2*A1leng*Gcut/pi+1.d0)
A2leng=ALAT*sqrt{ A2(1)**2 + A2(2)**2 + A2(3)**2 )
NRY=int(2*A2leng*Gcut/pi+1.d0)
A3leng=ALAT*sqrt{ A3(1)**2 + A3(2)**2 + A3(3)**2 )
NRZ=int(2*A1leng*Gcut/pi+1.d0)
Then rechoose NRX NRY NRZ to be near ODD numbers, but the'Prime numbers' should be avoided for the efficient FFT performance.
The file 'sym.C1' repersent that the calculated system has not symmetry and represent the identity matrix. The format is specific for FPSEID21 so it is recommended to obtaini this from sym.C1,
Here is an example of a script file to run 'cg_exe' which should be dependent on your Linux environment. One can see assignment for files at FORT41, FORT46, FORT42. (For 'Aurora' users, please use VE_FORT41, VE_FORT46, VE_FORT42 for file I/O settings.) These files are norm conserving pseudopotentials obtained by a scheme [N. Troullier and J. L. Martins, 'Efficient pseudopotentials for plane-wave calculation', Phys. Rev. B43, 1993 (1991)]. The pseudopotentials are in asci code, and downloadable from the box of the script below. Here, the manual recommends to create 'TR' directory on your account to store pseudopotentials in order to match with this script. Generally, the I/O number for the i-th type atom' (not the i-th atom) pseudopotentials is given 40+i when the atomic type corresponds to light elements, from H to Ne. On the other hand, when the i-th type atom corresponds to heavier elements, the pseudopotential files are separated into two with the I/O number 40+i and 40+i+5. This is very specific format and fits to FPSEID21 only. Note i-th atom's pseudopotential files with their names ending asr 'g.asci' or 'g.asc' should be used with I/O # 40+i. Meanwhile, file names ending as 'e.asci' or 'e.asc' should be used with I/O number 40+i+5. Indeed, one can confirm that rule in the following script, Pseudopotentials for Si and H atom can be downloaded when you click corresponding one displayed in a box below.
#$ -N cg_exe_Si111-H-1x1H
#$ -S /bin/bash
#$ -j y
#$ -e /home/youraccount/Si111-H/std_omp.err
#$ -o /home/youraccount/Si111-H/std_omp.out
#$ -pe mpi 16
export DIR=/home/youraccount
export DIR2=/home/youraccount/Si111-H
### --- input files
### pseudopotentials
export FORT41=$DIR/TR/TR.Si93g_asci ! Si Pseudopotentials (g)
export FORT46=$DIR/TR/TR.Si93e_asci ! Si Pseudopotentials (e)
export FORT42=$DIR/TR/TR.H99g_asc ! H Psedutopotentials
###
export FORT54=$DIR2/size.dat
export FORT55=$DIR2/sym.C1
###
export FORT20=$DIR2/rh.Si111-H ! input charge when 'CD=FILE' in 'Si111-H.in'
export FORT22=$DIR2/wf.Si111-H ! input wavefunction when 'WF=FILE' in 'Si111-H.in
### --- output files
export FORT23=$DIR2/wf.Si111-H_new ! output wavefunction (G-spave compact sphere)
export FORT88=$DIR2/wf_real.Si111-H ! output wavefunction (real space) used by 'sd_exe'
export FORT90=$DIR2/Vall.Si111-H
export FORT24=$DIR2/rh.Si111-H_new ! output charge density
export FORT25=$DIR2/Vint.temp
export FORT77=$DIR2/tau.Si111-H_new ! atomic coordinate stored at the end of 'cg_exe'.
export FORT78=$DIR2/need
export OMP_NUM_THREADS=4
cd $DIR2
$DIR/lm/cg_exe < Si111-H.in > Si111-H.out
When 'cg_exe' is performed, output files (binary) 'rh.Si111-H_new','wf.Si111-H_new', and 'wf_real.Si111-H_new' are generated. So please reneme 'rh.Si111-H_new as 'rh.Si111-H, ,'wf.Si111-H_new' as ,'wf.Si111-H', and 'wf_real.Si111-H_new' as 'wf_real.Si111-H'. Then, please check that the calculation reaches the SCF solution by typing
$ grep ITR Si111-H.out
to see whether convergency criterion is satisfied. If not, restart 'cg_exe' by changing the header of input file 'Si111-H.in' as follows; 'CD=ATOM' should be 'CD=FILE' and 'WF=INIT' should be 'WF=FILE. Then confirm the SCF congergency and rename all '*_new' files to those without the tail '_new'.'
The next step is running 'sd_exe'. To run 'sd_exe', visit manual for 'sd_exe'
One can also obtain the output file 'Si111-H.out' (asci) which is downloadable in above box. Here, description of some essencial parts is given.in red font as below.
-------
This is CG-code ...
-------
%%% Si(111)1x1 H-term cell %%%%
# OF COMMANDS = 20
NO. 1 COMMAND = STND OPERATION =
NO. 2 COMMAND = KPOINT OPERATION = MESH
NO. 3 COMMAND = ALAT OPERATION = 4.4427103214141699
- - - - - - - - - - -
- - - - - - - - - - -
NO. 19 COMMAND = MAXFN OPERATION = 0
NO. 20 COMMAND = OKSTEP OPERATION = 0.1D0
- - - - - - skipping - - - - -
INITIAL POSITION OF ATOM IN
1 0.0000000 0.0000000 2.2165260
2 4.1886275 0.0000000 3.6609519
3 4.1886275 0.0000000 8.0828717
4 -4.1886275 0.0000000 9.5397751
- - - skipping - - -
OMEGA= 2653.7234450
Read S-matrices fron file !!
# OF SYMMETRY OPERATIONS: NTOT = 1
S MATRICES: OP 1ST RAW 2ND RAW 3RD RAW
1 1 0 0 0 1 0 0 0 1
- - - - - skipping - - - -
A-VECTORS
6.2829412 6.2829412 0.0000000
-3.6274578 3.6274578 0.0000000
0.0000000 0.0000000 58.2184051
B-VECTORS
0.3535534 0.3535534 0.0000000
-0.6123724 0.6123724 0.0000000
0.0000000 0.0000000 0.0763111
- - - - skipping - - - -
**** MESHK: N = 4
TOTAL NO. OF K IN WHOLE BZ = 4
TOTAL NO. OF K IN WEDGE = 2
NO. COORDINATES WK
1 -0.2500 -0.2500 0.0000 1.0000
2 -0.2500 0.2500 0.0000 1.0000
- - - - skipping ----
*** ITR # 1 CONVERGENCE OF THE POTENTIAL IS ** 0.1365134D+00
*** ITR # 2 CONVERGENCE OF THE POTENTIAL IS ** 0.4149469D-01
- - - -
*** ITR # 23 CONVERGENCE OF THE POTENTIAL IS ** 0.2281881D-06
*** ITR # 24 CONVERGENCE OF THE POTENTIAL IS ** 0.9713493D-07
Store SCF local potential
Fourier components of eigen values
Fourier components of occupation #s
(See a column just below 'EIG').
IB
EIG
CONV'
SIN(ROT)
OCC
K VECTOR 1 -0.1767767 0.0000000 0.0000000 CARTESIAN
BAND (EV) 1 -0.1197297E+02 0.1811632E-07 0.1501256E-03 1.000
BAND (EV) 2 -0.1175938E+02 0.1691344E-07 0.1520060E-03 1.000
BAND (EV) 3 -0.1141360E+02 0.2046485E-07 0.1657217E-03 1.000
- - - - skipping - - -
K VECTOR 2 0.0000000 0.3061862 0.0000000 CARTESIAN
BAND (EV) 1 -0.1063678E+02 0.1632811E-08 0.1054169E-03 1.000
BAND (EV) 2 -0.1044811E+02 0.1177430E-08 0.1039971E-03 1.000
BAND (EV) 3 -0.1014525E+02 0.1802020E-08 0.1063807E-03 1.000
- - - - skipping - - -
****** TOTAL ENERGY: ETOT = -0.4867975154D+02 HR
-0.9735950309D+02 RYD
******
ESELF = 0.5052927104D+01 HR
******
EWA = 0.1842936419D+03 HR
******
ELOCAL = -0.4850683943D+03 HR
******
EXC = -0.1499663146D+02 HR
******
EH = 0.2317450508D+03 HR
******
EKINE = 0.1899942511D+02 HR
******
ENL = 0.1129422925D+02 HR
****** TOTAL FORCE: NEGATIVE (HARTREE/AU):
1 -0.0000203 0.0000032 0.0014520
2 0.0001712 0.0000095 -0.0002048
3 -0.0000624 -0.0000051 -0.0000924
- - - skipping - - -
13 -0.0005073 0.0000030 -0.0005740
14 0.0005073 -0.0000030 0.0005740