How to run?
Run 'tddft_exe' - the electron-ion dynamics under laser field
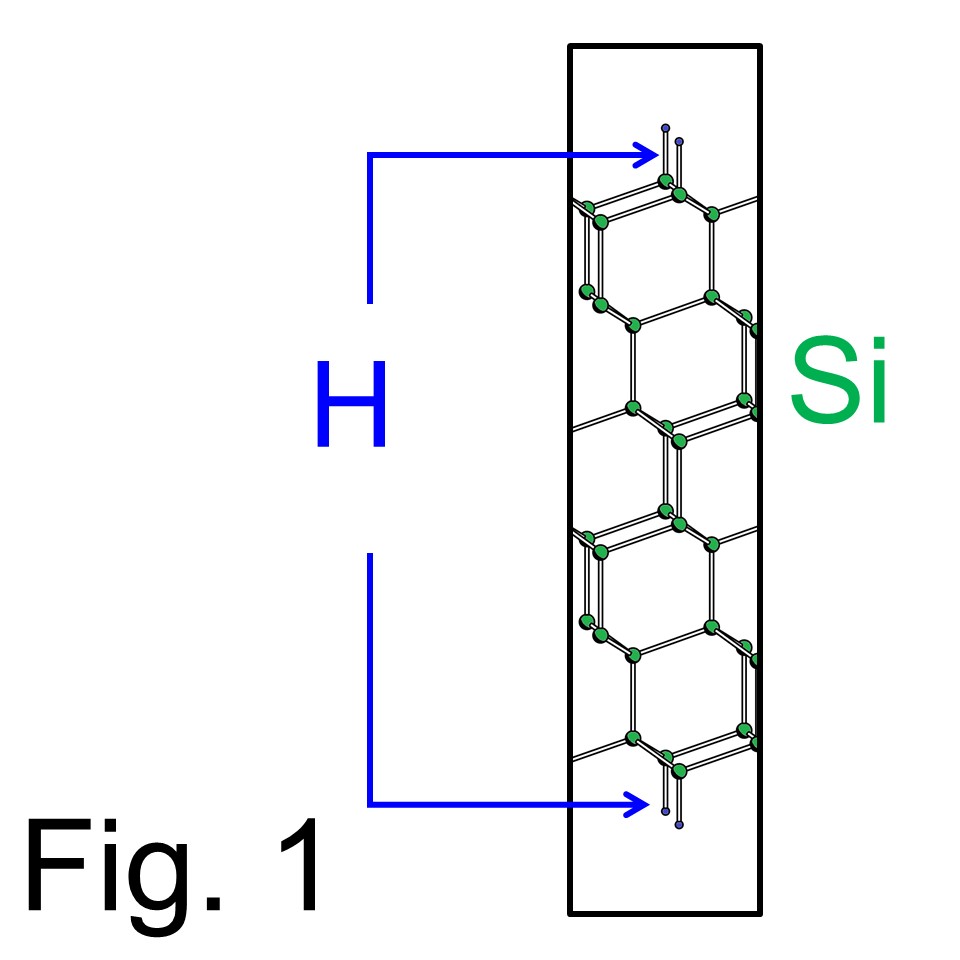 Here the manual steps to the final stage 'tddft_exe' with the example as illustrated in Fig. 1, which is a Si(111) slab model terminated by H atoms on top and bottom. In this manual, it is explained how to simulate laser induced H desorption from the Si(111) surface.
Here the manual steps to the final stage 'tddft_exe' with the example as illustrated in Fig. 1, which is a Si(111) slab model terminated by H atoms on top and bottom. In this manual, it is explained how to simulate laser induced H desorption from the Si(111) surface. Those who did not run 'cg_exe' or 'sd_exe' please back to manual pages. (To back 'cg_exe', click here. To back 'sd_exe', click here), Those who did not make executable modules 'cg_exe','sd_exe', and 'tddft_exe' should back to How to compile?.
The input data named as 'Si111-H_tm.in_1000steps' is displayed inside a box below. (This template file is obtained from download 'Si111-H_tm.in_1000steps') The changed parts from the 'Si111-H.in' are revision in header part, and addition of velocities and beginning time of TDDFT calculation at the bottom. The changed parts are shown in red font in the box below.
%%% Si(111)1x1 H-term cell %%%%
STND KPOINT=MESH ALAT=4.4427103214141699 ZVAL=50 RCUT=845 GCUT=12.0
PP=(KB,SOFT) CD=
OUTWF=23 METAL=(25,0) KCONT=0
RMIX=0.03 CONV=
1.4142135623730951 -0.8164965809277260 0.0000000000000000 A1 vector
1.4142135623730951 0.8164965809277260 0.0000000000000000 A2 vector
0.0000000000000000 0.0000000000000000 13.1042541360369356 A3 vector
2 Si12H2 # of Atomic types, chemical composition
12 2 28.0855 # of type 1 atoms, # of pseudo orbitals, atomic mass
0.0000000000000000 0.0000000000000000 0.0380725996019826 TAU( 1)
0.3333333333333333 0.3333333333333333 0.0628830683197726 TAU( 2)
0.3333333333333333 0.3333333333333333 0.1388370510257218 TAU( 3)
-0.3333333333333333 -0.3333333333333333 0.1638618412213989 TAU( 4)
-0.3333333333333333 -0.3333333333333333 0.2398223878715089 TAU( 5)
-0.0000000000000000 -0.0000000000000000 0.2642651909875856 TAU( 6)
-0.0000000000000000 -0.0000000000000000 -0.0380725996019826 TAU( 7)
-0.3333333333333333 -0.3333333333333333 -0.0628830683197726 TAU( 8)
-0.3333333333333333 -0.3333333333333333 -0.1388370510257218 TAU( 9)
0.3333333333333333 0.3333333333333333 -0.1638618412213989 TAU( 10)
0.3333333333333333 0.3333333333333333 -0.2398223878715089 TAU( 11)
0.0000000000000000 0.0000000000000000 -0.2642651909875856 TAU( 12)
-2 0 1.0 # of type 2 atoms (negative!), # of pseudo orbitals, atomic mass
-0.0000000000000000 -0.0000000000000000 0.3131888120278811 TAU( 13)
0.0000000000000000 0.0000000000000000 -0.3131888120278811 TAU( 14)
30.0 Rcut for LVGENX
4 total # of k-point mesh in one direction
1 1 0 starting points of k-point mesh for B1 B2 B3 vectors
2 2 4 skipping # of k-point mesh for B1 B2 B3 vectors
1 0 0 just these three lines
0 1 0
0 0 1
0 0 0
10 10 10 NDX NDY NDZ for DOS
2.0 2.0 for type 1 -- #s of electrons on s- and p-orbitals
1.0 0.0 for type 2 -- #s of electrons on s- and p-orbitals
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0
0.0 0.0 0.0 ! initial velocity of TAU ( 14)
0.0 ! begining tme (fs): or previous time in case of continuous run
Same as the input data for 'cg_exe' and 'sd_exe',
In the third line, CD=FILE means that the calculation starts with the valence charge density (the file is 'rh.Si111-H') obtained by 'sd_exe'. WF=32 means that the TDDFT calculation uses 32 bands for the valence wavefunctions. This is just for the case of Fig. 1. (Please note that the calculation by 'cg_exe' and 'sd_exe' used 32 bands as same as that used in 'tddft_exe'. However, in general cases, the 'tddft_exe' can provide the same MD results without including empty bands. Therefore, depending on available # of MPI processes, the value next to WF= can be modified as to maximize efficiency of parallel computing as 'tddft_exe' is parallelized w.r.t. the band index.
The next comping parameter is the iteration number for the SCF calculations, It was SCF=60 for running 'cg_exe' , but should be reduced as SCF=10 to avoid in wasting computation time in case of numerical bankruptcy. In the next line, FFTWF should be erased.
A new line starting with dt=0.40 is added here. dt is an interval of the time-step in atomic unit (1 a.u. = 0.0242 fs). tstep is number of steps of the simulation. TMOD is a step number at which the results are shown to the output file.
In th sixth line, the convergence criteria CONV=1.D-7 has been increased as CONV=1.D-5 for practical efficiency.
A new file'laser.dat' is necessary. The contents are shown in a box below. The first line is laser wavelength in 'nm' unit. The second line is a time in 'fs' unit, at when the laser intensity shows a peak, The third line is a half-value of the FWHM in unit of 'fs'. Here, as an example, the value is 1.0 thus means FWHN=2,0 fs. The last line denotes laser field vector at maximum intensity in the Cartesian coordinates in unit of V/angstrom.
800 ! wavelength in nm
3.5 ! peak position in fs
1.0 ! half of FWHM in fs
2.0d0 0.0 0.0 ! (Ex Ey Ez) in V/A
The script file to run 'tddft_exe' has some differences from that for 'cg_exe', which is assignments of new free-format asci files as follows:
'laser.dat'file at FORT53
'Eext'and 'Eext_new' files at FORT18 and FORT19, respectively
'Etot'and 'Etot_new' fiels at FORT28 and FORT29, respectively
'Avec' and 'Avec_new' files for FORT60 and FORT61, respectively
'Ework' and 'Ework_new' files for FORT61 and FORT62, respectively
At the first time-step of the TDDFT calculation, there should be initial values in 'Eext', 'Etot', 'Avec', and 'Ework' files.
'Etot' and 'Ework' should be
'Avec' should be
'Eext' should be
Indeed, 'Eext' and 'Eext_new' are dummy files but nees values in 'Eext' as above.
Here is an exmaple of the script file for running 'tddft_exe' in case of Fig. 1. The input data and out put data are downloadable from icons inside the box. Note the script is dependent on your Linux environment. The difference from the script for 'cg_exe' is hilighted by red fonts.
#$ -N tddft_exe_Si111-H-1x1H
#$ -S /bin/bash
#$ -j y
#$ -e /home/youraccount/Si111-H/std_omp.err
#$ -o /home/youraccount/Si111-H/std_omp.out
#$ -pe mpi 16
export DIR=/home/youraccount
export DIR2=/home/youraccount/Si111-H
### --- input files
### pseudopotentials
export FORT41=$DIR/TR/TR.Si93g_asci ! Si Pseudopotentials (g)
export FORT46=$DIR/TR/TR.Si93e_asci ! Si Pseudopotentials (e)
export FORT42=$DIR/TR/TR.H99g_asc ! H Psedutopotentials
###
export FORT53=$DIR2/laser.dat
export FORT54=$DIR2/size.dat
export FORT55=$DIR2/sym.C1
###
export FORT18=$DIR2/Eext ! Dummy (used for length Gauge)
export FORT19=$DIR2/Eext_new! Dummy (Used for length Gauge)
##
export FORT28=$DIR2/Etot ! ETOT in previous run
export FORT29=$DIR2/Etot_new ! ETOT at the end of run
##
export FORT60=$DIR2/Avec ! A-evctor in previous run
export FORT61=$DIR2/Avec_new ! A-vector at the end of run
##
export FORT62=$DIR2/Ework ! Work done by A-vector in previous run
export FORT63=$DIR2/Ework_new ! Work done by A-vector at the end of run
###
export FORT20=$DIR2/rh.Si111-H ! input charge
export FORT22=$DIR2/wf_fft.Si111-H ! input wavefunction
### --- output files
export FORT23=$DIR2/wf_fft.Si111-H_new ! output wavefunction (G-space full grid)
export FORT90=$DIR2/Vall.Si111-H
export FORT24=$DIR2/rh.Si111-H_new ! output charge density
export FORT25=$DIR2/Vint.temp
export FORT77=$DIR2/tau.Si111-H_new ! atomic coordinate stored at the end of 'cg_exe'.
export FORT78=$DIR2/need
export OMP_NUM_THREADS=4
cd $DIR2
mpirun -np 16 $DIR/lm/tddft_exe < Si111-H_tm.in_1000steps > Si111-H_tm.out_1000steps
In the bottom in above box, one can obtain the out put fiel 'Si111-H_tm.out_1000steps'. The beginning parts are very similar to the out put by 'cg_exe' but you will see display for parallel load disrtibution as follows
This is a load distribution for band indices.
nrest = 0
nbegin( 0 )= 1 nend( 0 )= 2
nbegin( 1 )= 3 nend( 1 )= 4
nbegin( 2 )= 5 nend( 2 )= 6
nbegin( 3 )= 7 nend( 3 )= 8
nbegin( 4 )= 9 nend( 4 )= 10
- - - - -
Then the output shows expectation values (energy levels), total energy, atomic positions, and velocities at every 'TMOD' steps (in current case at every 50 time steps) as follows;
Time (fsec) = 0.0000000000000000
**** expectation values (eV) ***
*** for 1 -th k-point
-0.12023403D+02 -0.11791839D+02 -0.11453088D+02 -0.10997574D+02
-0.10452258D+02 -0.99522407D+01 -0.75179199D+01 -0.70033570D+01
-0.62652226D+01 -0.55760218D+01 -0.50292431D+01 -0.47941576D+01
-0.46034832D+01 -0.43160955D+01 -0.40263948D+01 -0.36870239D+01
-0.31662306D+01 -0.31321113D+01 -0.29641639D+01 -0.26887333D+01
-0.25486855D+01 -0.23398649D+01 -0.19678424D+01 -0.18905517D+01
-0.17171152D+01 0.36664581D+00 0.59512272D+00 0.86551041D+00
0.12546349D+01 0.13379324D+01 0.16801698D+01 0.21815744D+01
*** for 2 -th k-point
-0.10698438D+02 -0.10492817D+02 -0.10197680D+02 -0.98049989D+01
-0.93356593D+01 -0.88388366D+01 -0.83778615D+01 -0.78888776D+01
-0.74958091D+01 -0.71478314D+01 -0.67999751D+01 -0.65165916D+01
-0.50201593D+01 -0.49191262D+01 -0.48927995D+01 -0.45944956D+01
-0.45456089D+01 -0.45003496D+01 -0.41974702D+01 -0.40396211D+01
-0.38409670D+01 -0.34446527D+01 -0.31935036D+01 -0.28628950D+01
-0.24255544D+01 0.98836510D+00 0.10001347D+01 0.18452992D+01
0.18752963D+01 0.19749452D+01 0.28162572D+01 0.29725999D+01
- - - - skipping - - -
****** TOTAL ENERGY: ETOT = -0.4877479647D+02 HR
-0.9754959294D+02 RYD
- - - skipping - - -
Time (fsec) = 0.0000000000000000
Positions
0.0000000000000000 0.0000000000000000 0.0380725996019826 TAU( 1)
0.3333333333333333 0.3333333333333333 0.0628830683197726 TAU( 2)
0.3333333333333333 0.3333333333333333 0.1388370510257218 TAU( 3)
-0.3333333333333333 -0.3333333333333333 0.1638618412213989 TAU( 4)
-0.3333333333333333 -0.3333333333333333 0.2398223878715089 TAU( 5)
0.0000000000000000 0.0000000000000000 0.2642651909875856 TAU( 6)
0.0000000000000000 0.0000000000000000 -0.0380725996019826 TAU( 7)
-0.3333333333333333 -0.3333333333333333 -0.0628830683197726 TAU( 8)
-0.3333333333333333 -0.3333333333333333 -0.1388370510257218 TAU( 9)
0.3333333333333333 0.3333333333333333 -0.1638618412213989 TAU( 10)
0.3333333333333333 0.3333333333333333 -0.2398223878715089 TAU( 11)
0.0000000000000000 0.0000000000000000 -0.2642651909875856 TAU( 12)
0.0000000000000000 0.0000000000000000 0.3131888120278811 TAU( 13)
0.0000000000000000 0.0000000000000000 -0.3131888120278811 TAU( 14)
Velocities
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 1)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 2)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 3)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 4)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 5)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 6)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 7)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 8)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 9)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 10)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 11)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 12)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 13)
0.0000000000000000 0.0000000000000000 0.0000000000000000 TAU( 14)
- - -
Time (fsec) = 9.1960000000000015
**** expectation values (eV) ***
*** for 1 -th k-point
-0.24527138D+01 -0.39673394D+00 0.10472097D+01 0.75329448D+00
-0.20642897D+01 -0.20677792D+01 0.22137208D+01 0.19345476D+00
-0.26362652D+01 -0.20601079D+01 -0.30559019D+01 -0.13652611D+01
-0.12113838D+01 -0.15826101D+01 -0.90888196D+00 -0.27295404D+01
0.12559438D+01 -0.47208109D+00 0.35544583D+01 0.37522232D+01
0.16571782D+01 0.25816898D+01 0.19239971D+01 0.21578943D+01
0.44989360D+01 0.98473958D+01 0.82103883D+01 0.10613458D+02
0.11618795D+02 0.14944322D+02 0.10081547D+02 0.13494579D+02
*** for 2 -th k-point
0.10582107D+01 -0.18649662D+00 -0.60454905D+00 0.61719284D+00
0.27785994D+01 0.25683293D+01 0.39228935D+01 0.14628025D+01
0.92880897D+00 0.81073229D+00 0.40032637D+01 0.43531474D+01
0.43549749D+01 0.19963713D+01 0.36127492D+01 0.42974925D+01
0.33817583D+01 0.39608561D+01 0.96274431D+00 0.50691312D+01
0.54842686D+01 0.52399215D+01 0.42655470D+01 0.18921902D+01
0.49305512D+01 0.13651843D+02 0.13411263D+02 0.14000486D+02
0.14191165D+02 0.15704823D+02 0.15367277D+02 0.14658990D+02
- - - - skipping - - -
****** TOTAL ENERGY: ETOT = -0.3284164019D+02 HR
-0.6568328038D+02 RYD
- - - skipping - - -
Time (fsec) = 9.6800000000000015
Positions
-0.0005738093867003 0.0007366905909603 0.0392532687075224 TAU( 1)
0.3341041320612999 0.3332131514856718 0.0616070163170294 TAU( 2)
0.3330736261373578 0.3317330378861919 0.1402805010908211 TAU( 3)
-0.3322623151358712 -0.3336655874357021 0.1616227306757774 TAU( 4)
-0.3319457870513178 -0.3344282105639471 0.2405077900306306 TAU( 5)
0.0002374647467333; 0.0002760103432352 0.2636980034777683 TAU( 6)
-0.0001447583589132 0.0006915932435568 -0.0390576468286915 TAU( 7)
-0.3319479465036790 -0.3341388421648399 -0.0612063296906515 TAU( 8)
-0.3333851730340425 -0.3329165303537411 -0.1398708271093426 TAU( 9)
0.3341309815513708 0.3328446050229766 -0.1612389999012748 TAU( 10)
0.3343704920870640 0.3352226469126762 -0.2406638248053374 TAU( 11)
0.0005017783877617 -0.0013999735694569 -0.2632865548263724 TAU( 12)
-0.0199830506390174 0.0020232932079802 0.3533172637448173 TAU( 13)
-0.0052540876581059 0.0193494191824069 -0.3521336900904135 TAU( 14)
Velocities
0.0000159411850188 0.0000336277889952 -0.0003257354076637 TAU( 1)
-0.0001109786641389 -0.0000105326342663 0.0004458158437197 TAU( 2)
-0.0000457098394316 0.0000152029824004 -0.0001432213334100 TAU( 3)
-0.0000293648969403 -0.0000078220001931 0.0001189474546982 TAU( 4)
-0.0000276166785918 -0.0000038944214707 -0.0002400483000792 TAU( 5)
-0.0000759301914792 0.0000124486983165 0.0006088376391160 TAU( 6)
-0.0000898088001837 -0.0000162087110865 0.0004355167728450 TAU( 7)
-0.0000280123366637 -0.0000084534726060 -0.0002870126165300 TAU( 8)
-0.0000516783069001 0.0000133118477902 0.0003118904526274 TAU( 9)
0.0000057093041024 0.0000042861541110 -0.0001067467223192 TAU( 10)
-0.0000039131068373 -0.0000264291146393 0.0002074250309303 TAU( 11)
0.0000621792542566 -0.0000668953737777 -0.0004528585673328 TAU( 12)
-0.0004241623097481 0.0003413296659772 0.0138013911474666 TAU( 13)
0.0003143999956733 0.0002136107588912 -0.0134692004392169 TAU( 14)
1000 steps took 0.156593824410D+04 sec
**** CPU TIME END OF PSPW: 1567.0915761 SEC
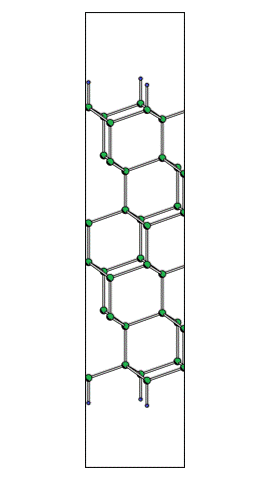
With use of time-evolution of atomic coordinates obtained from the output file 'Si111-H_tm.out_1000steps', one can make the animation like the one on the left. When you click this figure the animation starts. The yellow background indicates a moment when laser field is maximized. One frame of the movie corresponds around 1 fs.